
By Neha Lonkar
Chronic myelogenous leukaemia (CML) is a clonal hematopoietic stem cell disorder or simply put, a cancer of the blood-forming cells. CML is a disease that had wrecked the lives of those affected until the development of Gleevec, a drug that revolutionized the treatment of CML. (Iqbal & Iqbal, 2014)
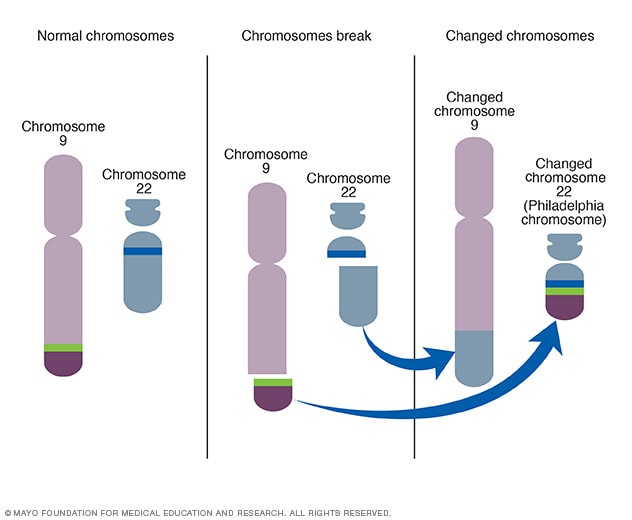
The development of Gleevec was preceded by several advancements in the understanding of the molecular biology of CML. These advancements included the discovery of a chromosomal abnormality eventually named the Philadelphia (Ph) Chromosome (Nowell and Hungerford,1960). Further research allowed us to understand that this abnormality was caused by a reciprocal translocation between chromosomes 22 and 9 to form a novel gene (Rowley,1973) that we now refer to as Bcr-Abl. A reciprocal translocation is the exchange of chromosomal material between 2 non-homologous chromosomes.

Figure 1: The Formation of Philadelphia Chromosome.
Source: How the Philadelphia chromosome forms. (n.d.).Mayo Clinic. Retrieved June 5, 2021, from https://www.mayoclinic.org/-/media/kcms/gbs/patientconsumer/images/2013/08/26/10/34/ds00564_im03579_c7_philadelphia_chromosomethu_jpg.jpg. Recreated by Aashna Sureka
Now, we know that genes code for proteins. Fusion genes such as Bcr-Abl code for different kinds of proteins called fusion proteins. Bcr-Abl codes for two fusion proteins, p185 and p210. The latter is seen in a higher percentage of CML patients (Druker et al., 1996). These proteins possess tyrosine kinase. This enzyme functions by transferring a phosphate group from ATP to tyrosine residues of proteins (Paul & Mukhopadhyay, 2004). Its activity is responsible for the incessant division of CML cells. How does tyrosine kinase cause unregulated proliferation of cells? It activates a signalling pathway called the Ras pathway (Salesse & Verfaillie, 2002).
Understanding this was a crucial point in the development of Gleevec because this drug aims to inhibit the activity of tyrosine kinase. Since ATP binding is pivotal to the function of kinases, any kinase inhibitors designed are aimed at targeting this ATP binding site.
Gleevec was discovered due to the combined efforts of scientists at Ciba Geigy (currently known as Novartis) who were searching for compounds that could function as kinase inhibitors and Brian Druker, an oncologist working on CML at the Oregon Health and Science University.
Gleevec started out as a compound called CGP 57148. This compound belongs to the 2-Phenylaminopyrimidine class of compounds and CGP 57148 was discovered through meticulous screening of these compounds in order to find one that could inhibit kinases. For simplicity, I will refer to CGP 57148 as CGP in this article.
Post identification, this compound went through a series of experiments and studies.
The first one of these was in vitro profiling using kinase assays. This was done to observe its effects on purified kinases. In vitro studies to investigate the effects of CGP on other processes such as induction of tyrosine phosphorylation by ligand binding to a transmembrane tyrosine kinase receptor or via cellular expression of a tyrosine kinase oncoprotein were also performed.
From these in vitro studies, CGP was understood to be an excellent inhibitor of the Abl protein tyrosine kinases with a good IC50 (the concentration of compound required to inhibit 50% activity of tyrosine kinase) value. The IC50 was considered good as it was low enough to not affect healthy cells.
Moreover, this compound showed high specificity and the authors did not see any notable inhibition of other protein kinases. There was one exception that showed a response and the kinase is known as platelet-derived growth factor (PDGF) receptor tyrosine kinase. However, it’s important to note that PDGF receptor sensitivity to the compound did not affect its ability to inhibit Bcr-Abl tyrosine kinase i.e., it was still highly specific.
Following this, experiments were conducted to determine the specificity with which the compound inhibited tumour formation. This experiment was performed on a strain of mice commonly used in cancer research (C3H/HeJ). These mice were inoculated with cells expressing either Bcr-Abl or v-Src genes. v-Src is a gene found in the Rous Sarcoma virus which is one of the viruses that cause cancer in chicken. Both Bcr-Abl and v-Src can form tumours in mice. However, when the compound CGP was allowed to act on these tumour forming cells, the authors observed that the compound only inhibited the growth of Bcr-Abl induced tumours and had no effect on v-Src induced tumours.
CGP was then studied to check for inhibition of cellular proliferation. Ordinary, healthy cells are growth factor-dependent. Growth factors promote cell division and when a cell becomes growth factor independent it proliferates uncontrollably. If cells express Bcr-Abl, their division becomes growth factor independent i.e., uncontrollable.
To check if CGP inhibited this, two cell lines were used. These cell lines were obtained from a murine factor-dependent cell line (32DcI3) and a human factor-dependent megakaryocytic cell line (MO7e). When these cell lines were modified to express p210Bcr-Abl they were known as MO7p210 and 32Dp210 (I will be referring to these modified cell lines as modified MO7 line and modified 32D line). This modification made them growth factor independent. All of these lines – modified and non-modified were used in the experiments.
The growth of these cell lines was observed under a variety of conditions in the presence/absence of both – growth factors and CGP. The authors observed that the non-modified cell lines were not inhibited by CGP in the presence of growth factors even with higher CGP concentrations of 10 µM. (See Figure 2 & 4)

Figure 2 – The graph indicates the number of viable cells of the non-modified cell line (MO7) when allowed to grow with varying concentrations of CGP.
Image Source: Druker, B. J., Tamura, S., Buchdunger, E., Ohno, S., Segal, G. M., Fanning, S., Zimmermann, J., & Lydon, N. B. (1996). Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr Abl positive cells. Nature medicine, 2(5), 561–566. https://doi.org/10.1038/nm0596-561. Recreated by Shruti S
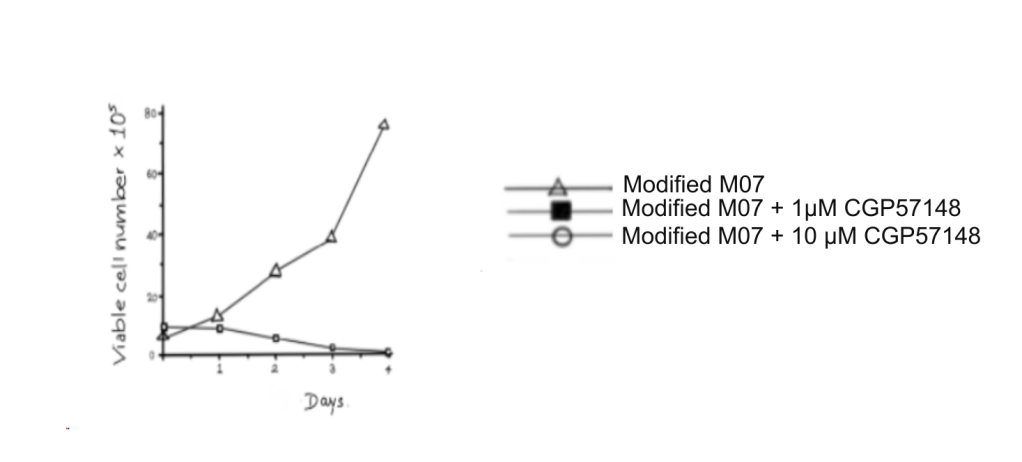
Figure 3: The graph indicates the number of viable cells of the modified MO7 cell line when allowed to grow with varying concentrations of CGP.
Image Source: Druker, B. J., Tamura, S., Buchdunger, E., Ohno, S., Segal, G. M., Fanning, S., Zimmermann, J., & Lydon, N. B. (1996). Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature medicine, 2(5), 561–566. https://doi.org/10.1038/nm0596-561. Recreated by Shruti S

Figure 4: The graph indicates the count of viable cells of non-modified cell line 32D.
Image Source: Druker, B. J., Tamura, S., Buchdunger, E., Ohno, S., Segal, G. M., Fanning, S., Zimmermann, J., & Lydon, N. B. (1996). Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature medicine, 2(5), 561–566. https://doi.org/10.1038/nm0596-561. Recreated by Shagun Soni.
However, the 2 factor-independent cell lines were inhibited by CGP regardless of the presence of growth factors.
The authors also observed that with the modified 32D cells the addition of Interleukin-3 (IL-3 is a growth factor) could rescue the cells to a certain extent but even with IL-3 most of the cells were inhibited. (Figure 5)

Figure 5: The graph indicates the count of viable cells of the modified 32D cell line (32Dp210).
Image Source: Druker, B. J., Tamura, S., Buchdunger, E., Ohno, S., Segal, G. M., Fanning, S., Zimmermann, J., & Lydon, N. B. (1996). Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature medicine, 2(5), 561–566. https://doi.org/10.1038/nm0596-561. Recreated by Shagun Soni.
Additionally, the authors noted that inhibition did not cause the cells to return to their growth factor-dependent state. On the other hand, a technique using antisense oligonucleotides (single-stranded DNA/RNA which is complementary to a specific sequence) allowed the cells to revert to their normal growth factor-dependent state. The authors theorised that this difference might’ve been because CGP only inactivates Bcr-Abl while the antisense strategy reduces the levels of Bcr-Abl by preventing gene expression.
Due to the fact that in modified MO7 cells, exposure to CGP resulted in cell death, the authors also hypothesized that Bcr-Abl could play a role other than inducing growth factor independence. The authors hypothesized that Bcr-Abl might play a role in the prevention of programmed cell death since cell death occurred on inhibition of Bcr-Abl tyrosine kinase activity.
Another experiment that the authors conducted was to study the inhibitory activities of CGP on colony formation. The inhibition of colony formation was checked in vitro using peripheral blood and bone marrow. Samples for this experiment were taken from both, patients who had CML and those that didn’t. To further check the specificity of CGP, bone marrow was taken from CML patients who had the Ph chromosome and one CML patient who didn’t have the Ph chromosome. Studying the data from these colony assays showed that CGP caused a decrease of Bcr-Abl colonies by 92-98%. The data also showed that bone marrow from non-CML patients was not inhibited at low levels of CGP. At higher levels of the compound such as 10µM, there was about a 15-20% inhibition.
These studies concluded that only cells which had a permanently active Bcr-Abl protein tyrosine kinase would be affected by CGP.
The authors also highlighted the differences and advantages of using CGP over antisense oligonucleotide strategy. The major advantage lies in the specificity of this compound which prevents it from affecting normal marrow.
In addition to the above-mentioned points, CGP showed very low, if any, toxicity in animals. This low level of toxicity and high selectivity of CGP made it an excellent choice for use in in vivo therapy against CML and any other leukaemias which are Bcr-Abl positive and this is exactly what the authors hoped for.
In about 2 years, a clinical study using this compound was initiated by Druker. CGP was renamed STI571. The clinical study showed an incredible result. Complete hematologic responses (CHR refers to the return of normal blood cell counts) were seen in almost every patient and more than half the patients treated showed cytogenetic responses i.e., reduction in the number of cells containing the Ph chromosome (Druker et al., 2001). Druker and his team launched a study 60 months after their last clinical trial to assess the long-term effects of the compound. The survival rate was incredibly high. However, the drug has to be used continuously. Any discontinuation results in relapse, even in patients with undetectable levels of Bcr-Abl (Druker et al., 2006).
Yet, even if we take this factor into consideration, Gleevec or Imatinib, as it’s now known, was a game-changer in drug therapy against cancer. It was a drug designed to inhibit a specific protein that was vital to the pathogenicity of a specific disease. Brian Druker, Nicholas Lydon and Charles Sawyer won the Lasker-DeBakey Clinical Medical Research Award in 2009 for their work on this drug. Their work on Gleevec was followed by the creation of a number of kinase inhibiting drugs aimed at treating CML as it completely transformed science’s understanding of cancer therapy.
Did you enjoy reading this? Let us know what you thought of this article in the comments section below!
ABOUT THE AUTHOR
Neha Lonkar
She is an undergraduate student studying Microbiology and Biochemistry at St. Xavier’s College, Mumbai.
We would like to thank Dr. Seema Das, HOD, Department of Life Science and Biochemistry, St Xavier’s College for reviewing this article and for her valuable inputs.
The Boffin Bloggers
MAIN REFERENCE:
Druker, B. J., Tamura, S., Buchdunger, E., Ohno, S., Segal, G. M., Fanning, S., Zimmermann, J., & Lydon, N. B. (1996). Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature medicine, 2(5), 561–566. https://doi.org/10.1038/nm0596-561
REFERENCES:
- Druker, B. J., Guilhot, F., O’Brien, S. G., Gathmann, I., Kantarjian, H., Gattermann, N., Deininger, M. W., Silver, R. T., Goldman, J. M., Stone, R. M., Cervantes, F., Hochhaus, A., Powell, B. L., Gabrilove, J. L., Rousselot, P., Reiffers, J., Cornelissen, J. J., Hughes, T., Agis, H., . . . Larson, R. A. (2006). Five-Year Follow-up of Patients Receiving Imatinib for Chronic Myeloid Leukemia. New England Journal of Medicine, 355(23), 2408–2417. https://doi.org/10.1056/nejmoa062867
- Druker, B. J., Talpaz, M., Resta, D. J., Peng, B., Buchdunger, E., Ford, J. M., Lydon, N. B., Kantarjian, H., Capdeville, R., Ohno-Jones, S., & Sawyers, C. L. (2001). Efficacy and Safety of a Specific Inhibitor of the BCR-ABL Tyrosine Kinase in Chronic Myeloid Leukemia. New England Journal of Medicine, 344(14), 1031–1037. https://doi.org/10.1056/nejm200104053441401
- How the Philadelphia chromosome forms. (n.d.). Mayo Clinic. Retrieved June 5, 2021, from https://www.mayoclinic.org/-/media/kcms/gbs/patient-consumer/images/2013/08/26/10/34/ds00564_im03579_c7_philadelphia_chromosomethu_jpg.jpg
- Iqbal, N., & Iqbal, N. (2014). Imatinib: A Breakthrough of Targeted Therapy in Cancer. Chemotherapy Research and Practice, 2014, 1–9. https://doi.org/10.1155/2014/357027
- Nowell, P. C., & Hungerford, D. (1960). A Minute Chromosome in Human Chronic Granulocytic Leukemia. Science, 132, 1497. https://doi.org/10.1126/science.132.3438.1488
- Paul, M. K., & Mukhopadhyay, A. K. (2004). Tyrosine kinase – Role and significance in Cancer. International Journal of Medical Sciences, 101–115. https://doi.org/10.7150/ijms.1.101
- ROWLEY, J. D. (1973). A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia identified by Quinacrine Fluorescence and Giemsa Staining. Nature, 243(5405), 290–293. https://doi.org/10.1038/243290a0
- Salesse, S., & Verfaillie, C. M. (2002). BCR/ABL: from molecular mechanisms of leukemia induction to treatment of chronic myelogenous leukemia. Oncogene, 21(56), 8547–8559. https://doi.org/10.1038/sj.onc.1206082

{kind=link}
{kind=link}